Theory
Investigators
- Stefan Brackertz
Description - Why do we need research on molecular physics?
Molecular physics is quantum mechanics as developed in the beginning of 20th century. Quantum mechanics is about setting up the Schroedinger equation and solving it. So why do we need any research in moecular physics? Why do we need complicated experiments? Why do we need people dealing with the theory of molecular physics? Why don't we just solve the Schroedinger equation and are done?
The answer is: Already for the smallest molecules the Schroedinger equation is much too complicated to be solved. And even with nowadays supercomputers there is no chance to directly solve the Schroedinger equation in a brute-force manner.
The common approach to overcome this problem is to replace the full Schroedinger equation by an easier model which could be solved analytically or at least numerically. These models are inspired by symmetry considerations and typically involve some parameters which have to be determined by experiments. This comes along with three challenges:
- Develop a model appropriate for the molecule under the interesting conditions
- Measure the molecule
- Link model and measurement cogently
But why don't we just settle for the measurements?
Approaches
Symmetries
If a physical description is to complicated the first thought is often to try an approximation appropriate to your question (i.e. you're only interested in a certain regime or in a specific feature of the system). However, this is just the second best approach. Using symmetry to simplfy your description is preferable as you avoid the errors of an approximation. In fact, it is very common to do both, i.e. choose polar coordinates for a problem with rotation symmetry resulting in an equation of motion for two angles instead of three carthesian coordinates and then do an approximation of the still challenging equation of motion for the angles. Numeric simulations as i.e. done in our carbon cluster subgroup typically also require to simplfy the problem using symmetries first.
In view of this, our goal is to not only use the obvious symmetries like rotations, but also involve representation theory (a subarea of mathematics) to make use of "hidden symmetries".
Floppy Molecules
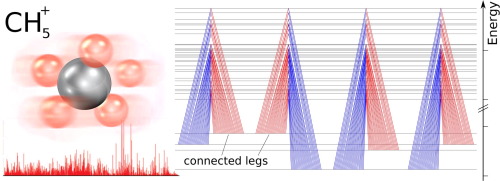
A particular challenge are "floppy molecules": The models common in molecular physics have already been invented in the 1930s and rely on the assumption that molecules have a fixed geometry with fixed bond lengths, angles and so on – just as you have painted them in school. In fact, for a lot of molecules like water, CO2 and alcohol this approach is rather successful, at least as long as you keep them cold. In contrast, ammonia, He-H3+, CH5+ and similar molecules do not have a fixed geometry. The troublemaker of ammonia is the famous, but still comparatively easy umbrella flip-motion which can be incorporated by a fix of the common model. But for completely floppy molecules like CH5+ there is no easy trick; indeed this molecule has eluded any description until we understood a few years ago the exact reason why the common approaches fail for this molecule.
Dealing with floppy molecules and symmetries is not only a business on its own
How far can we get without a model?
In the advent of quantum mechanics people measured atomic and molecular spectra and had no clue what they observed. In these day Ritz invented a combinatorial principle to nevertheless reconstruct the states of hydrogen from the measured transitions. In 2015 our group brought out this old approach again to evaluate the measurements of CH5+ for which no model existed at all and none had a clue what we measured. This evaluation was an important foundation of the recent approaches to develop models for this molecule. It was also the foundation to further develop this principle to a computer program allowing for the semi-automatic reconstruction of the states of a molecule from its transition energies without further preknowledge.
Our aim is to make this tool not only helpful for the development of models of molecules for which no approprpriate models exist so far. Altough there already exist a lot of tools like pgopher or the Loomis-Wood Plot software developed by our absorption subgroup we also want to use the combinatorial approach to make life easier with ordinary molecules allowing for an assignment of model and measurement based on states and experimental selection rules instead of transitions as today.
Further approaches
Selected Publications
-
Searching for new symmetry species of CH5+ – From lines to states without a model
S. Brackertz, S. Schlemmer, O. Asvany
J. Mol. Spectrosc. 342, 73-82 (2017) -
Unifying the rotational and permutation symmetry of nuclear spin states: Schur-Weyl duality in molecular physics
H. Schmiedt, P. Jensen, S. Schlemmer
J. Chem. Phys. 145, 074301 (2016) -
Rotation-vibration motion of extremely flexible molecules – The molecular superrotor
H. Schmiedt, P. Jensen, S. Schlemmer
Chem. Phys. Lett. 672, 34-46 (2017)
Acknowledgments
This project has been supported via DFG project AS 319/2-2, DFG project SCHL 341/6-2 and DFG SFB 1601.